
Hi, and welcome to this video about amyotrophic lateral sclerosis (ALS).
Amyotrophic lateral sclerosis, commonly known as Lou Gehrig’s disease, is an incurable, fatal, progressive neurodegenerative disease. ALS primarily affects motor functions, causing those affected to experience both upper and lower motor neuron signs and symptoms.
What Are Neurons?
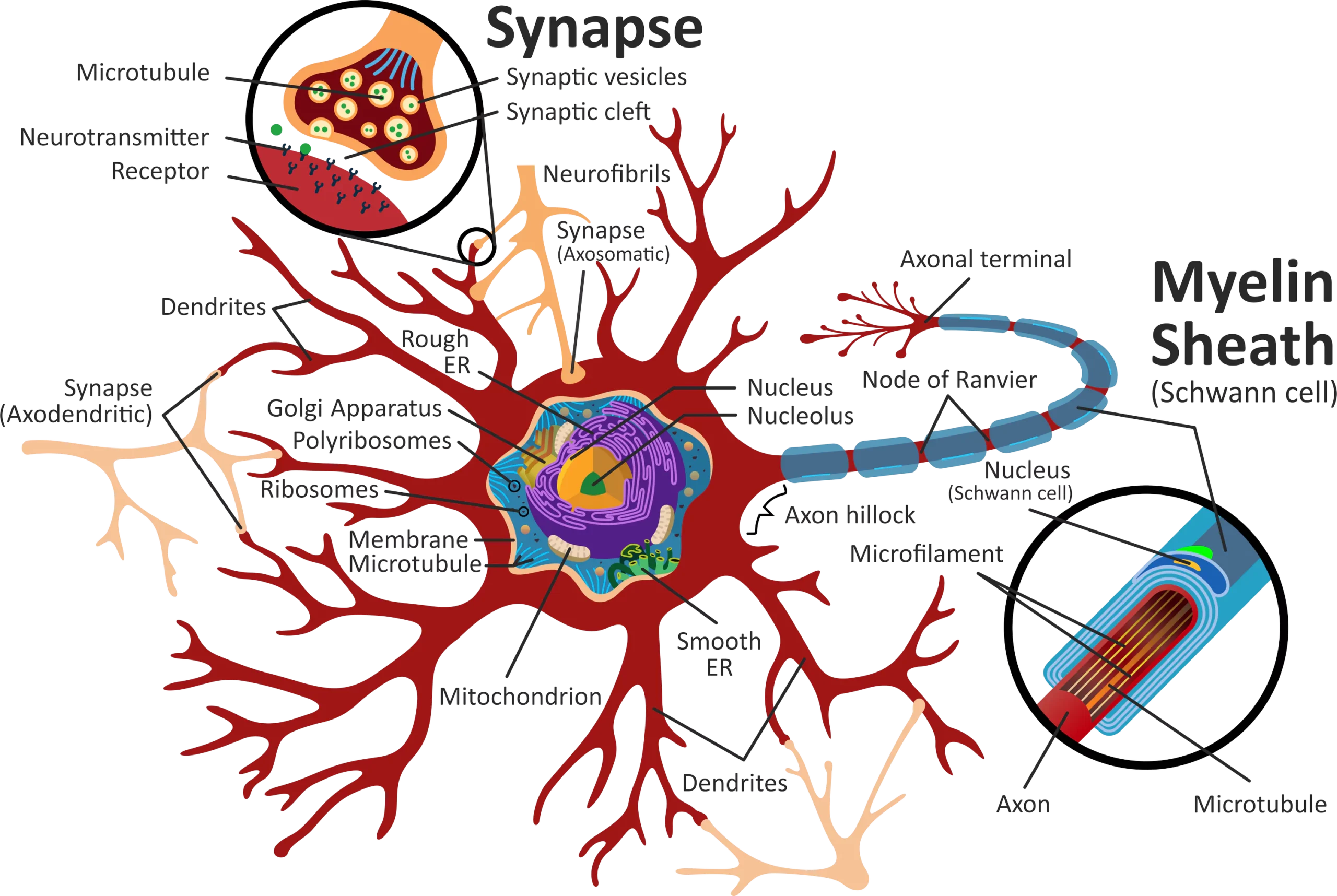
Before we talk about the effects of ALS, let’s talk about the parts of a neuron:
- The cell body, which controls the functions of the cell
- The dendrites, which carry impulses to the cell body
- The axon, which carries impulses away from the cell body
- The myelin sheaths, which protect the axon and facilitate transmission of electrical impulses
- The axon terminals, which release transmitters so that impulses can pass from one neuron to another
Motor Neurons
Upper motor neurons are those in the central nervous system that allow for voluntary movement and provide muscle tone. Upper motor neurons transmit impulses to the lower motor neurons. Signs of upper motor neuron dysfunction include increased tone, spasticity, and hyperreflexia, but with reflexes preserved in atrophied muscles.
Lower motor neurons connect to the central nervous system and carry impulses to and from muscles and glands. Signs of lower motor neuron dysfunction include weakness, wasting, fasciculations (or twitching), and an absence of reflexes.
With ALS, the cell bodies begin to die and the axon and myelin sheaths begin to degenerate and scar so that impulses cannot be transmitted, resulting in muscle atrophy. ALS destroys both upper and lower motor neurons.
ALS Causes
While ALS typically begins in one part of the body, it spreads to encompass the entire motor system, resulting in increasing weakness and paralysis of all voluntary muscles and eventually includes those muscles required for talking, eating, and breathing.
Onset of ALS usually occurs in people who are in their 50s and 60s, although some physicians have reported seeing that cases have increased in younger people over the last two decades. The reason for this is unknown, although some of the increase in findings may be due to enhanced diagnostics and medical technology.
There are many different theories about the cause of ALS, including oxidative stress from free radicals that damage cells, mitochondrial abnormalities, autoimmune response, and genetics.
Ninety to ninety-five percent or more of ALS cases are classified as sporadic, with the remaining cases classified as familial. Males are twice as likely to develop the sporadic form as females in individuals younger than 70 years of age. Prevalence becomes more even across both sexes as age increases above 70.
Five to ten percent of cases are genetic and linked to mutations in chromosome 9, so these cases are classified as familial or genetic. These same mutations are associated with frontotemporal disease, so some patients may develop both ALS and frontotemporal disease, which is characterized by personality changes, apathy, language impairment, and weakness in the arms and legs.
ALS Risk Factors
In most cases of ALS, the cause is simply unknown; however, possible risk factors may include:
- Smoking (according to increased evidence)
- Exposure to environmental toxins (such as those encountered by veterans fighting in the Gulf War)
- Viral infections
- Trauma
- Diet
- Behavioral factors
- Autoimmune diseases
Life expectancy for most types of ALS is 3 to 5 years. In fact, 80 percent of patients die within 5 years. Respiratory failure is the most common cause of death. With more slowly progressing forms, some can live 10 or 20 years or, in rare cases, longer.
For example, Stephen Hawking, the famous theoretical physicist, was diagnosed in 1963 at age 21, needed a wheelchair by the late 1960s, lost speech in the 1970s, and required a tracheostomy and ventilation in 1985. Nonetheless, Hawking lived 55 productive years after the diagnosis, dying in 2018.
ALS Presentation
ALS is variable in presentation, depending on the type of onset, which is referred to as the phenotype. Bulbar, cervical, and lumbar onsets are most common, but within these types, initial symptoms may be primarily upper motor neuron, lower motor neuron, or mixed, so the clinical picture may be quite different.
- Bulbar onset involves symptoms primarily of the face, mouth, and jaw, so people experience dysphagia, slurred speech, drooling, and—over time—breathing difficulties. Their voices may sound harsh and they may have difficulty articulating and experience fasciculations of the tongue.
- Cervical onset impacts the back of the head, upper back, neck, shoulders, and arms, resulting in weakness, stiffness, cramping of fingers, and wrist drop.
- Lumbar onset affects the legs, causing weakness, stiffness, difficulty walking, and foot drop, resulting in a “slapping” gait.
Most cases present with the cervical or lumbar phenotype, 20 to 25 percent with bulbar, and a small percentage with a less common phenotype.
Flail Arm and Flail Leg Syndromes
Flail arm and leg syndromes are less common phenotypes that can be confused with the cervical or lumbar phenotypes initially, although they do not include hypertonia or clonus.
While these phenotypes are also progressive, they progress more slowly than other phenotypes.
- Flail arm syndrome is a lower motor neuron disorder that involves progressive weakness and muscle wasting beginning with the proximal muscles of the upper extremities.
- Flail leg syndrome is a lower motor neuron disorder of the lower extremities and involves progressive weakness and wasting beginning with distal muscles of the lower extremities.
Respiratory Onset
Respiratory onset is another rare form. This onset usually presents with increasing dyspnea on exertion as well as at rest (orthopnea). Respiratory onset has a worse prognosis than other forms because it begins with respiratory problems, the most fatal complication of ALS.
Regardless of the phenotype, eventually symptoms progress through four stages to include virtually the entire body:
- Stage one is the beginning, characterized by the type of onset. Muscles begin to weaken, and the person may experience cramping, twitching, and increased fatigue.
- Stage two is the middle stage, during which the muscles most affected become paralyzed and other muscles begin to stiffen and weaken. Painful contractures tend to develop and joints become rigid and deformed. The person will have increased difficulty swallowing and may experience labile emotions. Patients are at risk of developing aspiration pneumonia from increased drooling.
- Stage three is the late stage. At this point, 90 percent or more of the muscles are paralyzed and the person is no longer able to swallow effectively and must be fed through a feeding tube, such as a percutaneous endoscopic gastrostomy. The patient will have increased difficulty breathing and will need help for all personal needs. The person will have difficulty speaking and may experience headaches and dizziness.
- Stage four is the ending stage, during which the person will need intubation and ventilation to breathe and a feeding tube for nourishment. Some may opt for a tracheostomy to prolong life for a time, while others allow the disease to progress more naturally to death.
Between 20 and 50 percent of people with ALS exhibit a pseudobulbar affect, which results in sudden and involuntary bouts of inappropriate emotional expression, such as crying or laughing, with crying more common than laughing. This response may last for several minutes and is not associated with the person’s actual emotional state.
ALS-Functional Rating Scale-Revised (ALSFRS-R)
The Revised ALS-Functional Rating Scale is commonly used to evaluate change in functional status for those with ALS. The scale assesses 12 different items in 4 domains for a maximum of 48 points. Each item is scored from 0 to 4, with 4 being a normal finding.
- The first domain is bulbar, which assesses speech, salivation, and swallowing.
- The second domain is fine motor, which assesses handwriting, ability to cut food (with or without a gastrostomy), and dressing and hygiene.
- The third domain is gross motor, which assesses the ability to turn in bed, walk, and climb stairs.
- The fourth and last domain is respiratory, which assesses dyspnea, orthopnea, and respiratory insufficiency.
People’s rate of decline may vary, with some, for example, losing 1 point a month and others up to 6 points.
ALS Treatment
Treatment for ALS is primarily supportive and may include:
- Occupational therapy to help handle functional changes
- Physical therapy to help to maintain muscle tone and reduce contractures
- Psychological counseling to help deal with the diagnosis and prognosis of an incurable disease
- Insertion of a feeding tube or percutaneous endoscopic gastrostomy
- Non-invasive or invasive ventilation, including a tracheostomy
- Mobility aids, such as braces, wheelchairs, and lifts
- Communication aids, such as computerized programs and speech synthesizers
The FDA has approved two medications specifically for treatment of ALS:
The first drug is riluzole (brand name Rilutec®), which extends the time to tracheostomy and life expectancy, but only for a few months. Riluzole is available in tablet form and in an oral suspension and is taken twice daily.
The second drug is edaravone (brand name Radicava®), which slows the decline in function by about 33%. Edaravone must be administered intravenously, usually daily for 14 days followed by a 14-day drug-free period and then the cycle repeats.
Additional medications to manage symptoms may include
- Diazepam or baclofen to reduce muscle spasticity
- Lorazepam to reduce anxiety
- SSRIs to reduce depression
- Amitriptyline or trihexyphenidyl to reduce saliva and drooling
- Gabapentin and NSAIDs to reduce pain
- Anticholinergics, such as scopolamine and glycopyrrolate, to reduce drooling
- Dextromethorphan and quinidine to control the pseudobulbar affect
Diagnosis is often delayed because initial symptoms are nonspecific to ALS, so diagnosis is often arrived at through a process of elimination of other possible diseases.
Unfortunately, there is still much about ALS that is not fully understood, but research is ongoing in the search for a cure or medications to better slow the advance of the disease. Clinical trials are testing a number of different drugs, some now in phase 2 trials.
That’s all for this review. Thanks for watching, and happy studying!
